Diágnostico de la acromegalia [1]
El diagnóstico de esta enfermedad requiere confirmación bioquímica, ya que es clínico. La acromegalia se sugiere por el trastorno típico del paciente relacionado con el agrandamiento progresivo de la región acral y la modificación de la apariencia facial.
En esta nota
- Ensayos de GH
- Límite de GH para el diagnóstico
- Medición de IGF-1
- Pruebas de estimulación
- Situaciones clínicas difíciles
- Evaluación tumoral y funcional de la hipófisis
- Estudios de imagen
- Investigaciones funcionales de la hipófisis
- Tratamiento
- Pronóstico y resultado
Como hemos mencionado en artículos anteriores, el diagnóstico de acromegalia se basa en la presencia de concentraciones elevadas de la hormona del crecimiento (GH) que no disminuyen después de una prueba de tolerancia a la glucosa oral (PTGO). La confirmación del diagnóstico se obtiene al observar un aumento en la concentración sérica del factor de crecimiento similar a la insulina tipo 1 (IGF-1), ajustado por la edad. El IGF-1 es el principal factor de crecimiento que depende de la GH.
Es importante tener un alto índice de sospecha para considerar la acromegalia (ver “¿Qué es la acromegalia? [2]”), ya que el diagnóstico puede retrasarse varios años desde el inicio de los síntomas. Este retraso se asocia con una mayor mortalidad y comorbilidades, destacando la importancia de la detección temprana y el tratamiento oportuno.
Diferentes grupos de pacientes requieren evaluación para la acromegalia:
- Pacientes jóvenes: El crecimiento lineal excesivo durante la infancia o adolescencia debe ser evaluado para descartar un exceso de GH.
- Pacientes adultos: Se debe considerar la acromegalia en adultos con agrandamiento acral, rasgos faciales sugestivos o síntomas asociados a la enfermedad. Estos incluyen:
- Dolor de cabeza frecuente
- Transpiración excesiva.
- Hipertensión.
- Apnea del sueño Oligomenorrea.
- Artralgias.
- Síndrome del túnel carpiano.
- Diabetes mellitus tipo 2
El análisis de fotografías de los pacientes puede ser útil para detectar la acromegalia en aquellos con rasgos faciales sutiles [1].
Ensayos de GH
Los ensayos de GH han evolucionado desde los RIA (radioinmunoensayo) de baja sensibilidad. Con el tiempo se introdujeron los ensayos radioinmunométricos de anticuerpos de dos sitios (IRMA), no competitivos, que ofrecían una mayor sensibilidad. Actualmente, los ensayos más utilizados son los de anticuerpos no isotópicos de dos sitios, algunos de los cuales están automatizados. Las diferencias en las metodologías de los ensayos pueden explicar la variabilidad en los resultados de GH, la cual circula en el plasma en diversas formas moleculares, lo que también contribuye a esta variabilidad.
La estandarización con el IS 98/574 busca alinear las mediciones. Anteriormente, se utilizaban diversas preparaciones estándar de GH para calibrar los ensayos. Esta estandarización busca uniformizar las mediciones de GH y facilitar la comparación de resultados.
Límite de GH para el diagnóstico
Aunque un nivel plasmático basal de GH elevado sugiere acromegalia, también se pueden encontrar concentraciones elevadas en personas sanas debido a la fluctuación natural de la GH.
- Es apropiado realizar mediciones basales de GH e IGF-1 ante la sospecha de acromegalia.
- Si la GH es inferior a 0,4 μg/l y el IGF-1 es normal, se descarta la acromegalia.
- Si la GH es superior a 0,4 μg/l o el IGF-1 está elevado, se realiza una PTGO.
- Si el valor mínimo de GH durante la PTGO (nadir) es inferior a 1 μg/l, se descarta la acromegalia.
- Si el nadir de GH permanece por encima de 1 μg/l, se confirma la acromegalia.
- Con los ensayos de alta sensibilidad actuales, se ha propuesto reducir el límite de positividad de la PTGO a 0,3 μg/l.
Medición de IGF-1
El nivel de IGF-1 aumenta en paralelo con la concentración de GH y debe medirse ajustado por la edad, ya que disminuye con el tiempo. Factores como el embarazo, la pubertad y el período pospuberal se asocian con niveles elevados de IGF-1. La proteína IGFBP3, que transporta el IGF, también suele estar elevada en pacientes con acromegalia, pero proporciona poca información adicional para el diagnóstico.

 [3]
[3]  [4]
[4]  [5]
[5]
Pruebas de estimulación
Hasta un 50% de los pacientes tienen un aumento en su concentración de GH después de la estimulación con la hormona liberadora de tirotropina (TRH) y/o la hormona liberadora de gonadotropina (GnRH). Sin embargo, estas pruebas no tienen valor diagnóstico, ni tampoco la respuesta a la GHRH.
Situaciones clínicas difíciles
Existen casos atípicos y complejos que pueden surgir en el diagnóstico de la acromegalia, donde las pruebas estándar no son concluyentes o no se ajustan al cuadro clínico.
- Nadir de GH bajo con IGF-1 alto: Algunos pacientes con signos claros de acromegalia y niveles elevados de IGF-1 pueden presentar un nadir de GH menor a 1 μg/l durante la PTGO. Esto plantea un desafío diagnóstico, ya que el valor de corte tradicional para confirmar la acromegalia es 1 μg/l. En estos casos, algunos autores sugieren que, con el uso de ensayos de GH altamente sensibles, el límite de positividad de la PTGO debería reducirse a 0,3 μg/l.
- Acromegalia resuelta espontáneamente: En otros casos, pacientes con un cuadro clínico típico de acromegalia pueden presentar concentraciones normales de IGF-1 y GH. Esta situación podría indicar una remisión espontánea de la enfermedad, posiblemente debido a una necrosis del adenoma hipofisario.
- Condiciones especiales: En ciertas situaciones como la diabetes mellitus, insuficiencia renal crónica, embarazo o pubertad, la medición de GH y/o IGF-1 puede no ser confiable para el diagnóstico o la evaluación del tratamiento. Estos escenarios requieren un enfoque individualizado y la consideración de otros parámetros clínicos.
- Pseudoacromegalia: Algunos pacientes presentan características acromegaloides con niveles normales de GH e IGF-1. Esta condición se denomina pseudoacromegalia y en algunos casos se asocia a una resistencia grave a la insulina. El diagnóstico diferencial en estos casos es fundamental para evitar tratamientos innecesarios.

Evaluación tumoral y funcional de la hipófisis.
Una vez confirmado el diagnóstico de acromegalia, es fundamental realizar una evaluación completa antes de comenzar el tratamiento (ver “Tratamiento de la acromegalia [6]”). Esta evaluación debe enfocarse tanto en las características del tumor como en la funcionalidad de la glándula pituitaria para obtener un panorama detallado de la condición del paciente.
Algunos efectos tumorales locales son los siguientes:
- Dolor de cabeza: Es frecuente, típicamente retroorbitario o frontal, causado por el adenoma y el exceso de GH.
- Trastornos visuales: Suele haber una compresión del quiasma óptico por el adenoma, resultando en alteraciones del campo visual, desde la pérdida de visión periférica temporal hasta hemianopsia bitemporal e incluso ceguera.
- Otras manifestaciones: Extensión del tumor:
- Supraselar: hidrocefalia por obstrucción del agujero de Monro.
- Infraselar: lisis del suelo selar e invasión del seno esfenoidal, con riesgo de rinorrea del líquido cefalorraquídeo.
- Lateral: compresión de pares craneales III, IV, V o VI por invasión del seno cavernoso, con riesgo de epilepsia focal por afectación del lóbulo temporal.
Estudios de imagen
Las técnicas utilizadas para evaluar la presencia y características del adenoma hipofisario en pacientes con acromegalia han ido cambiando con el tiempo:
- Radiografía de silla turca: Aunque antes se usaba para detectar agrandamiento de la silla turca, desmineralización o erosión, ahora se considera obsoleta. En pacientes con tumores muy grandes, la radiografía podía mostrar la desaparición completa del contorno de la silla turca, mientras que los tumores de crecimiento asimétrico producían un doble contorno en la vista lateral o una oblicuidad del suelo selar en la vista frontal.
- Resonancia magnética (RM): Ha ido reemplazado a la tomografía como el examen neurorradiológico de elección para pacientes con adenomas hipofisarios, especialmente en casos de acromegalia ya que ofrece una mejor visualización de los tejidos blandos y permite una evaluación detallada del tamaño, la forma y la ubicación del adenoma.
- Microadenomas (< 10 mm): Se ven como anomalías redondeadas, bien delimitadas y homogéneas en la RM. Suelen ser hipointensos en T1, pero a veces isointensos, dificultando su detección. El contraste con gadolinio ayuda a visualizarlos mejor.
- Signos indirectos: Asimetría de la hipófisis, abultamiento, elevación del diafragma selar o desviación del tallo hipofisario pueden indicar un microadenoma, incluso si no se ve directamente.
- Macroadenomas (> 10 mm): Generalmente son isointensos en T1 sin contraste, pero hiperintensos tras la administración de gadolinio.
- Evaluación de la extensión tumoral: La RM también permite evaluar la extensión del adenoma en las estructuras circundantes:
- Extensión supraselar: Se observa si el tumor comprime, desplaza o afecta al quiasma óptico.
- Extensión infraselar y lateral: Se evalúa la invasión del seno esfenoidal y del seno cavernoso. La invasión del seno cavernoso se confirma si la arteria carótida interna está rodeada por el tumor.
- Secreción ectópica de GHRH: La ausencia de un adenoma visible en la RM, o la presencia de una hipófisis hiperplásica abultada, sugiere que la acromegalia puede estar causada por una secreción ectópica de GHRH, es decir, la producción de GHRH por un tumor fuera de la hipófisis.
Investigaciones funcionales de la hipófisis
El adenoma en la fosa selar puede comprimir la hipófisis sana o el tallo hipofisario, afectando la secreción normal de hormonas. Esta compresión puede generar una deficiencia de gonadotropinas, causando disfunción sexual en los hombres y trastornos menstruales en las mujeres. También puede afectar la producción de tirotropos, con niveles bajos de T4 y normales de TSH (hormona estimulante de la tiroides). El déficit de corticotropos se evalúa midiendo los niveles de cortisol plasmático matutino o mediante pruebas hormonales, como metirapona o CRH (hormona liberadora de corticotropina), dependiendo del protocolo de cada centro.
Tratamiento
Los objetivos principales del tratamiento para la acromegalia son aliviar los síntomas (ver “Síntomas de la acromegalia [7]”), reducir el tamaño del tumor hipofisario, prevenir la recurrencia y mejorar tanto la morbilidad como la mortalidad a largo plazo. Las investigaciones han permitido definir con mayor precisión lo que se considera una "cura" o buen control de la enfermedad, fijando la concentración de GH por debajo de 2 μg/l y normalizando los niveles de IGF-1. Una estrategia de tratamiento gradual, que incluye cirugía, radioterapia y/o terapia médica, es clave para lograr estos objetivos (figura 1).
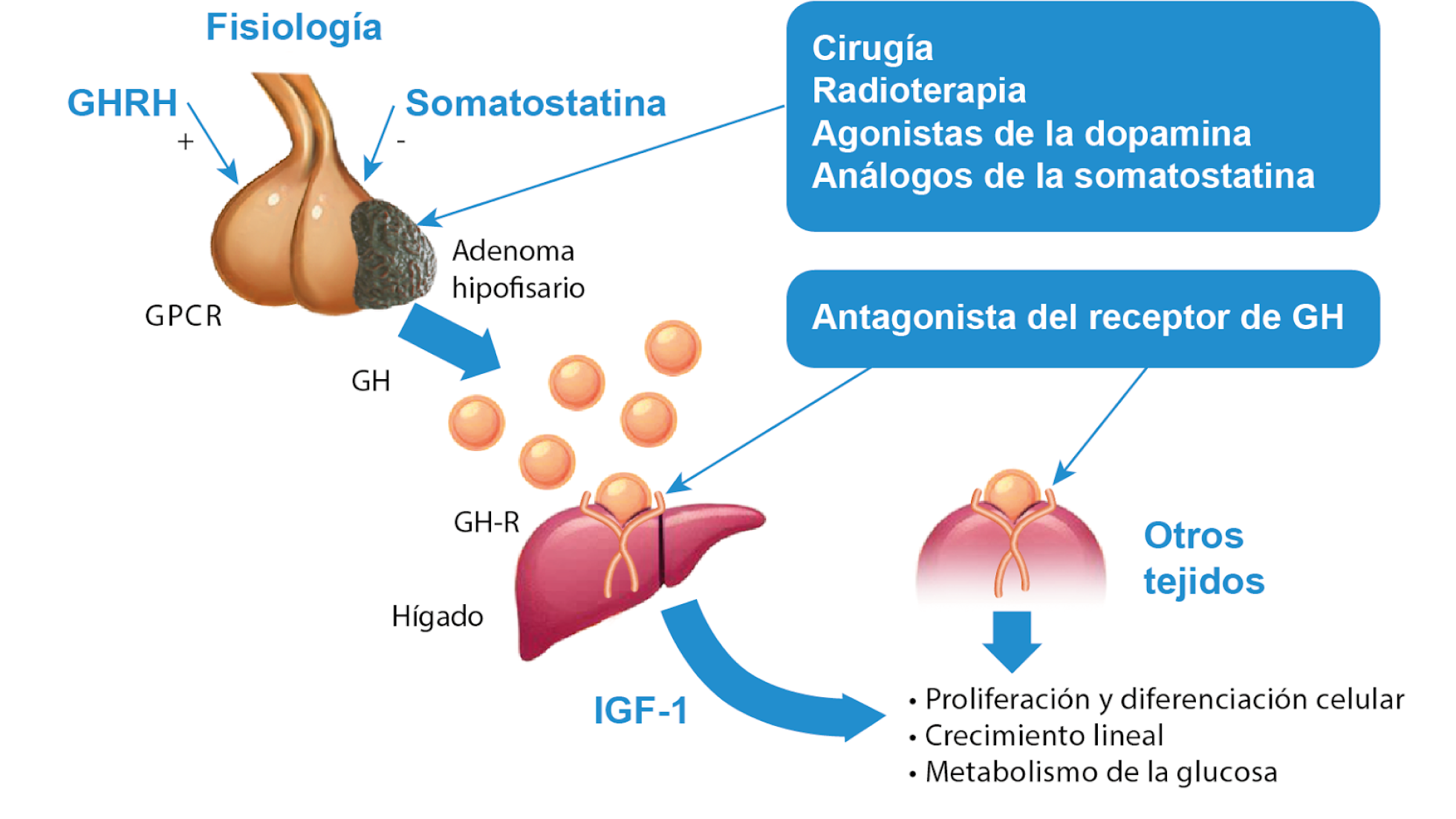
Figura 1: Los diferentes tratamientos para la acromegalia actúan en diversas áreas. La cirugía, la radioterapia, los análogos de la somatostatina y los agonistas de la dopamina trabajan directamente sobre el adenoma hipofisario, que es la causa principal de la secreción excesiva de GH. Por otro lado, los antagonistas de los receptores de la GH funcionan bloqueando el receptor de esta hormona en los tejidos periféricos, lo que impide que la GH ejerza sus efectos en esos tejidos.
La cirugía
Este suele ser el tratamiento de primera elección para la acromegalia. La extirpación del tumor, generalmente a través de la vía transesfenoidal, es la forma más rápida de reducir los niveles de GH e IGF-1, aunque estos solo se normalizan en el 40% al 70% de los casos, dependiendo del tamaño del tumor y las concentraciones preoperatorias de GH. Las técnicas endoscópicas son comúnmente utilizadas en centros especializados. Si la cirugía no es exitosa o no es una opción, se consideran la radioterapia y tratamientos farmacológicos como alternativas.
Radioterapia
La radioterapia se dirige al tumor hipofisario con el objetivo de reducir la producción de GH y normalizar los niveles de IGF-1.
Tipos de radioterapia:
- Radioterapia externa fraccionada: Este es el tipo más común de radioterapia para la acromegalia. Consiste en administrar una dosis total de radiación de 50 Gy en aproximadamente 25 sesiones diarias.
- Radioterapia estereotáctica ("radiocirugía"): Esta técnica, que incluye métodos como el bisturí gamma, administra una dosis alta de radiación de forma muy precisa al tumor, minimizando el daño a los tejidos sanos circundantes.
Eficacia de la radioterapia:
- La radioterapia fraccionada puede normalizar los niveles de GH (<2 μg/l o 6 mUI/l) e IGF-1 en un 5% a 60% de los pacientes después de un seguimiento promedio de 7 años.
- Estudios con seguimientos más largos indican que la radioterapia fraccionada puede normalizar el IGF-1 en más del 70% de los pacientes después de 10 años.
- La radiocirugía estereotáctica parece tener una eficacia similar a la radioterapia fraccionada, con menos del 20% de los pacientes logrando niveles normales de IGF-1 y GH <2 μg/l después de 4 años.
- La concentración inicial de GH antes de la radioterapia puede influir en la probabilidad de éxito del tratamiento.
Efectos secundarios y limitaciones:
- La radioterapia puede causar insuficiencia de la hipófisis anterior en un 80% a 100% de los pacientes después de 10 a 15 años.
- Aunque la radionecrosis y la neuropatía óptica son raras en la actualidad, existe un riesgo de accidente cerebrovascular que puede manifestarse muchos años después del tratamiento.
- La radiocirugía estereotáctica está reservada para pacientes con tumores pequeños situados al menos a 5 mm del quiasma óptico.

Tratamiento médico
Agonistas de la dopamina
- La bromocriptina, un agonista de la dopamina, puede aliviar los síntomas y reducir las concentraciones de GH, pero solo normaliza los niveles de IGF-1 en un pequeño porcentaje de pacientes (10%).
- La cabergolina, otro agonista de la dopamina, parece ser más eficaz que la bromocriptina en el control de la acromegalia.
Análogos de somatostatina
- Mecanismo de acción: Los análogos de la somatostatina, como la octreotida y la lanreotida, inhiben la secreción de GH al unirse a los receptores de somatostatina (SST) en las células somatotropas del adenoma.
- Preparaciones disponibles:
- Octreotida (Sandostatin®): Puede ser inyectado por vía subcutánea (SC), generalmente por el propio paciente, a una dosis de 100 a 200 μg dos o tres veces al día. Este fue el primer análogo de este tipo que se comercializó, en la década de 1980, y representó un verdadero avance terapéutico. Octreotida LAR (Sandostatin® LAR 10–20 o 30 mg) es la versión de liberación sostenida de octreotida y se administra por vía intramuscular, una vez al mes. El tratamiento generalmente se inicia con la dosis mediana y luego se ajusta (disminuye o aumenta) según la concentración de GH. Alternativamente, es posible aumentar o disminuir la frecuencia de las inyecciones.
- Lanreotida (Somatuline® Autogel®): Está disponible para inyección subcutánea profunda cada 28 días, en dosis de 60, 90 y 120 mg.
- Eficacia: Los análogos de la somatostatina pueden normalizar los niveles de GH en un 60% a 70% de los pacientes y los niveles de IGF-1 en un 50% a 80% de los pacientes.
- Efectos antitumorales: Además de su efecto antisecretor, los análogos de la somatostatina también pueden reducir el volumen del tumor en un 20% a un 70% de los pacientes.
- Uso como terapia de primera línea: En casos seleccionados, como contraindicaciones para la cirugía o tumores invasivos, los análogos de la somatostatina pueden usarse como primera opción de tratamiento.
- Desventajas: Los análogos de la somatostatina requieren tratamiento continuo, pueden tener efectos secundarios gastrointestinales, pueden causar cálculos biliares y son costosos.
Antagonista del receptor de GH: pegvisomant
- Mecanismo de acción: El pegvisomant (Somavert®) actúa a nivel periférico, bloqueando la acción de la GH en sus órganos diana al unirse a los receptores de GH e impedir su dimerización, inhibiendo así la producción de IGF-1.
- Administración: Se administra por vía subcutánea, con ajuste de dosis según la respuesta hormonal (normalización del IGF-1).
- Eficacia: El pegvisomant es altamente eficaz para normalizar los niveles de IGF-1 en más del 90% de los pacientes.
- Uso actual: Actualmente se reserva para pacientes en los que los análogos de la somatostatina no son efectivos.
- Monitoreo: Es necesario monitorear el volumen del tumor durante el tratamiento con pegvisomant, ya que se ha observado un pequeño aumento en algunos pacientes.
- Efectos adversos: Los efectos adversos son raros y generalmente leves, incluyendo aumento de las transaminasas y, en casos excepcionales, hepatitis.
Estrategia terapéutica actual
La estrategia terapéutica actual para la acromegalia sugiere evaluar las ventajas, desventajas y costos de cada tratamiento (figura 2). Tras la cirugía, si la enfermedad persiste, se prioriza el uso de análogos de la somatostatina antes que la radioterapia. Si estos fallan, se puede reconsiderar una reintervención si quedan restos tumorales significativos, o bien el uso de pegvisomant antes de recurrir a la radioterapia. El tratamiento médico debe evaluarse anualmente, y si la cirugía o radioterapia están contraindicadas, el tratamiento con somatostatina de primera línea es una opción viable.

Figura 2: Una posible estrategia para el manejo actual de la acromegalia. Referencias: AS (análogos de la somatostatina); GHRA (antagonista del receptor de GH, pegvisomant). Fuente del gráfico: Adaptado de “Acromegaly Symptoms Review”; Philippe Chanson and Sylvie Salenave. Acromegaly. Review. Orphanet Journal of Rare Diseases 2008, 3:17 doi:10.1186/1750-1172-3-17
Pronóstico y resultado
La evolución a largo plazo de los pacientes con un tratamiento adecuado puede mejorar notablemente tanto la esperanza como la calidad de vida.
Sin tratamiento:
- La acromegalia sin tratamiento conlleva una mayor mortalidad, disminuyendo la esperanza de vida en aproximadamente una década.
- Las principales causas de muerte en pacientes no tratados son (ver “Causas de la acromegalia [8]”):
- Enfermedades cardiovasculares (60%)
- Complicaciones respiratorias (25%)
- Cáncer (15%)
- Los accidentes cerebrovasculares también representan una causa frecuente de muerte, especialmente en mujeres. Es importante destacar que muchos estudios previos incluyeron pacientes tratados con métodos ya obsoletos, como la craneotomía y la radioterapia convencional, que podrían haber contribuido a la mortalidad observada.
Impacto del tratamiento:
- El tratamiento eficaz de la acromegalia ha revolucionado el pronóstico de la enfermedad.
- La concentración de GH tras el tratamiento es el factor que mejor predice la supervivencia, independientemente de la causa de muerte.
- Si se logra controlar la secreción de GH (<2 μg/l, o <5 mUI/l, o normalización del IGF-1), la esperanza de vida se equipara a la de la población general.
Factores pronósticos:
- Factores de buen pronóstico:
- Control de la secreción de GH (<2 μg/l o normalización del IGF-1)
- Factores de mal pronóstico:
- Concentraciones elevadas de GH/IGF-1
- Hipertensión arterial
- Miocardiopatía
Calidad de vida:
- La acromegalia impacta negativamente en la calidad de vida.
- Un tratamiento eficaz mejora la calidad de vida, aunque no siempre la restablece por completo.
Perspectivas actuales:
- Gracias a las estrategias terapéuticas actuales, la mayoría de los pacientes con acromegalia logran un buen control de la secreción de GH/IGF-1 y no presentan complicaciones asociadas al crecimiento tumoral.
- La utilización de criterios más estrictos para definir la curación, junto con un manejo proactivo de las comorbilidades, ha mejorado considerablemente el pronóstico de la acromegalia (ver “Manejo de la acromegalia [9]”).
- No obstante, incluso con un control adecuado de la enfermedad, pueden persistir secuelas como dolor articular, deformidades y una calidad de vida mermada.
En resumen, la acromegalia no tratada se asocia a una mayor mortalidad, pero el tratamiento eficaz puede normalizar la esperanza de vida y mejorar la calidad de vida. El control de la secreción de GH es crucial para un buen pronóstico. Aunque se han logrado avances significativos en el tratamiento, algunos pacientes pueden experimentar secuelas a largo plazo, lo que subraya la importancia del diagnóstico precoz y la intervención terapéutica oportuna.